
One of the increasingly popular forms of cloud computing are so-called SaaS services, or Software as a Service. This solution is based on the assumption that applications and databases are installed on the provider’s servers, and access to applications is usually provided through a website. The most popular applications of this type include: OneDrive, SalesForce.com, Google Apps, Concur, and Dropbox. The advantage of such an approach is that the Customer gets access to solutions with specific functionalities, without having to invest in IT infrastructure. An additional advantage is that such programs can be accessed through both desktops and mobile devices.
Cloud solutions are also becoming an increasingly popular tool in pharmaceutical companies, including areas deemed as critical to good manufacturing practices (GMP). When we talk about software supporting a critical GMP area, we must of course remember that such a solution will require validation. How can such validation be carried out correctly? What are the potential risks? What kind of cooperation can you expect between the pharmaceutical company and the provider? These are the principal questions, that will be addressed in this article.
Definition of requirements
As in any other case, we should start by preparing the User Requirement Specification (URS). However, when choosing a SaaS solution, it should be kept in mind that apart from functional requirements describing system operation or quality requirements covering GMP aspects, we should particularly consider the issues of data management and data integrity.
We must first of all acknowledge that the same solution is already in use or may be used in the future by other companies, including our competitors. This is important because the data will be stored on external servers managed by an external company. If the system is to additionally process personal data, we are entering the area of personal data protection regulations. Therefore, it will be important to specify the requirements in this area in accordance with the internal security and data management policy. When creating the URS, we should at least pay attention to a few of the following issues:
– server location
– dedicated server
– archiving and backup
– access management
– updates, system corrections
– testing
The above issues do not, of course, exhaust the whole range of questions and requirements that can be posed to the SaaS system, but they should help to point the way when writing the URS.
Provider Audit
According to the requirements of Annex 11 of the GMP, the IT systems provider must be audited. In the case of cloud solutions, this is particularly important because we entrust the provider with the implementation of the business process, the system and its care, as well as data and its security. It may also be one of the few opportunities to analyse the provider’s quality system in detail.
Satisfactory responses of the provider to the requirements defined in the URS must be verified and confirmed at this stage. To this end, the provider must be verified with regard to whether they have appropriate procedures in place, whether these procedures are implemented and applied in practice, and whether their staff is adequately trained. From the perspective of the client – a pharmaceutical company – one of the key points of the audit should be the verification of validation documentation and, if the provider does not have such documentation, then the verification of technical and test documentation. Another key audit point should be the aspects related to system security, starting from the verification of system architecture, through encrypted communication protocols, to the verification of penetration tests.
It should be kept in mind that according to GMP regulations, the final responsibility for the business process always falls to the pharmaceutical company, even when the process has been delegated to the provider as a part of the Software as a Service solution. Taking this into account, the provider audit is a critical element of the solution validation process.
Software as a Service solution validation process
As a rule, the SaaS solution validation process should not differ significantly from the validation of other computer systems owned by a pharmaceutical company. Therefore, if the company has defined validation procedures in this area, they should be applied. Below is an example of one of the possible approaches. An example of a validation process is presented in the diagram below:
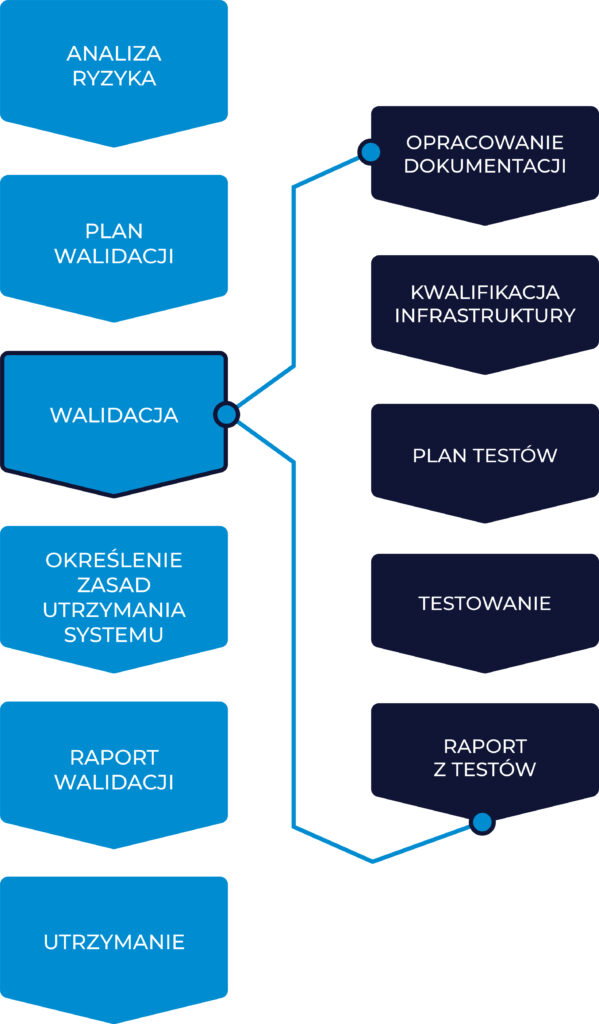
Risk Analysis – the risk analysis process should commence with the start of the validation process, and all subsequent activities (qualification and its scope, scope of tests, required documentation, etc.) should be planned and performed based on a “risk-based approach”. Various tools can be used for risk analysis, some of the most popular are FMEA and Expert Assessment, which will also work well in this case.
Validation Plan – based on the conducted risk analysis and the results of the provider’s audit, a validation plan should be developed, which should take into account all the key stages of the process, while also taking into account the specificity of the SaaS solution. This mention of the provider’s audit result is not a coincidence, because the conclusions of the audit will strongly influence which actions should be planned for implementation on their own, which actions should be given to the provider for implementation, and which actions can be omitted because they are already being implemented by the provider.
Documentation Preparation – the pharmaceutical company is responsible for the development of the User Requirements Specification, while other technical documentation (Functional Specifications, Technical Specifications, description of system architecture, description of configurations, interfaces and others) should be developed and provided by the software provider. In order to verify that the provider’s documentation properly covers the URS, the preparation of the Traceability Matrix can be started at this stage. The Traceability Matrix is part of the pharmaceutical company’s validation documentation and is designed to demonstrate that user requirements have been implemented in the system (the connection between the URS and the corresponding Functional Specification) and that they have been properly tested (the connection between the URS and the corresponding tests).
Infrastructure Qualification – validation tests must be carried out on qualified infrastructure. The software provider should at this stage provide qualification documentation for servers (at least a test and production server). Without confirmation of infrastructure qualification status, validation tests should not be started.
Performing validation tests – the scope of tests, approach to testing, reporting rules and error assessment criteria as well as the method of documentation should be described in the test plan. The tests can be divided into two groups: IT Tests (Unit Tests, Source Code Review, System Tests, Integration Tests, Security Tests) and Business Tests (User Acceptance Tests, End-to-End Tests). The provider is responsible for the technical part of the system, so if the audit results in the testing area were positive, then in the case of IT Tests, it is possible to refer to tests performed by the provider. It will be important to verify the available test documentation and the scope of testing. Such an approach significantly reduces the amount of testing on the part of the pharmaceutical company, because only Business Tests will be performed. After the tests are completed, the Traceability Matrix should be completed in order to show that each of the requirements has been properly tested. Finally, a Test Report summarising the results should be prepared. All errors reported during testing and their current status should be verified. Errors deemed as critical and important should be resolved before the Test Report is finalised.
Determination of System Maintenance Rules – after completion of all tests and prior to finalization of the Validation Report, system maintenance rules should be established and described. If necessary, appropriate procedures and instructions should be developed and training provided. These actions should also be summarised in the Validation Report and detailed maintenance policies described in a separate Service Level Agreement. Validation Report Preparation – at the end of the validation process a Validation Report should be prepared with a summary of the entire process. If any discrepancies or deviations occurred during the validation process, they should also be described and evaluated. If any discrepancies have not been resolved at this stage, the risk reduction measures implemented and the deadline for resolving open discrepancies should be presented. The Validation Report should include a statement that the object of validation has been approved and released for use in production. All documentation that is produced as part of the validation process must be prepared and approved before signing the Validation Report. This also applies to training materials, procedures, instructions, etc.
System Maintenance – Service Level Agreement
A validated system working within the SaaS solution, like any other, requires maintenance from both the technical and the validation sides. By system maintenance we mean a number of activities carried out by the provider in order to ensure the correctness and continuity of system operation. These activities include: monitoring system operation, making changes, corrections and updates, testing, managing permissions and access, archiving and backup, incident management, providing support lines (helpdesk, 2nd and 3rd lines of support) and others.
From the point of view of the pharmaceutical company, it will be crucial to continuously maintain the validated status, implemented in the change control process, which should combine aspects related to:
– incident and defect management – how defects are handled by the 1st, 2nd and 3rd support line, who opens the incident and when, what the communication channels between the customer and the provider are
– risk assessment – what the assessment criteria are, what needs to be done in case of an emergency notification, how the level of risk translates into further implementation steps, e.g. testing
– document management – which documents and how they should be updated
– testing – how the range of tests is selected, what the testing methods are, how the tests are documented
– training – whether correction or system updates are changing the way the system works and operates, user training is required
This process, in simple terms, should comply with GMP requirements, i.e.:
– formal change requests should be available – this applies to bug fixes, when an incident may be reported, and updates
– each change request should be assessed at least in terms of its impact on the system, documentation and testing; on this basis, an implementation plan should be drawn up together with a list of necessary actions to be taken
– the implementation should be monitored on an ongoing basis in terms of both substantive and qualitative correctness
– finally, the change should be assessed as to whether it has been implemented correctly
Since the system is maintained by the provider and it is the provider who is responsible for the implementation of processes such as, for example, change control, it is crucial that there is a formal service level agreement between the pharmaceutical company and the provider, which will specify the above issues, both in terms of the content of these processes and the principles of cooperation (communication channels, response times, access to documentation, etc.).
Summary
Due to their numerous advantages, SaaS (Software as a Service) cloud solutions are increasingly popular among pharmaceutical companies, also in GMP critical areas. And although the validation required in these cases can be challenging, it seems that the key aspect of the whole process is the choice of the provider, preceded by a detailed audit. However, such a conclusion should not surprise anyone. The quality of the system and its security in this case depends primarily on the provider, their awareness of the GMP requirements and the quality processes being implemented properly. However, it should be kept in mind that when deciding on a given solution, a pharmaceutical company is responsible for the whole validation process and should know how it wants to carry out such validation before deciding on such a solution.






